The 2024 International Plant & Animal Genome Conference (PAG 31)
For over three decades, the Plant and Animal Genome Conference (PAG) has been a meeting known for fostering collaboration, and showcasing groundbreaking advancements in the fields of plant and animal genomics. Established in 1993, PAG has evolved into a conference that converges experts, emerging talents, and innovators in genomics. This year’s PAG 31 is set to take place from January 12-17, 2024, in San Diego, CA, USA.
This year, the conference will include the following plenary speakers: Appolinaire Djikeng (Centre for Tropical Livestock Genetics and Health), Scott Edwards (Harvard University), Lucy van Dorp (University College of London), Katrien M. Devos (University of Georgia), Amy Marshall-Colón (University of Illinois Urbana-Champaign), Dirk Inzé (VIB-UGent Center for Plant Systems Biology), Virginia Walbot (Stanford University). Notable among them is Scott Edwards from Harvard University, who will lead the Plenary Session on “Comparative Population Pangenomes: A New Frontier for the Evolutionary Analysis of Birds.” Edwards’ expertise in evolutionary biology promises an enlightening exploration into avian genomics, unveiling new frontiers in our understanding of bird populations and their evolution.
The Scientific Programs at PAG 31 present diverse workshops, each dedicated to unraveling distinct facets of genomics. From decoding biodiversity to exploring the intricacies of genome annotation, attendees can expect a comprehensive dive into a wide array of workshops. If you are attending this year’s meeting, here is a list of sessions you might not want to miss.
Day 1, Jan 12, 2024 (Fri)
Vertebrate Genomes Project: Completing Phase 1: Led by Erich Jarvis, this session will give an overview of Phase 1 of the Vertebrate Genomes Project (VGP). The VGP project aims to generate near error-free reference genome assemblies of ~70,000 extant vertebrate species. The goal for phase 1 is to create a chromosome level assembly for 260 vertebrate orders. A talk by Andreas Pfenning from Carnegie Mellon University will highlight the session. His talk will describe how the genomes from the ordinal lineages sequenced in phase 1 are used to detect signatures of genome sequence evolution. Specifically, he will compare coding and noncoding sequences in the genome and how they evolve.
ORG.one Genomes for Conservation of Critically Endangered Species – ORG.one is a pilot-stage initiative sponsored by Oxford Nanopore designed to expedite the sequencing of critically endangered species. Through this project, biologists are able to rapidly sequence species near the individual’s habitat without harming or sacrificing the endangered individual. This workshop will highlight the latest developments in organizational initiatives, sequencing facilities, and the creation of de novo reference genomes. Discussions will delve into recent progress in nanopore sequencing, genome assembly, annotation, and their practical use in conservation genomics. Aziz Ebrahimi of Purdue University will present his research on the white walnut tree. Gabrielle Hartley of the O’Neil lab at the University of Connecticut will be giving a talk on the Eastern Hoolock Gibbon and the rapid karyotype evolution of small apes.
Animal Genomics and Adaptation to Climate Change: This workshop will provide insight on the implications of climate change for animal agriculture, spanning extreme weather, drought, heatwaves, and shifts in pathogen distribution. These elements significantly impact food security and animal farming across both well-equipped and under-resourced global regions. Understanding the genomic and physiological responses of animals facing climate change is pivotal in devising strategies to counter its adverse effects. This enables the use of tactics like breeding climate-resilient animals and adopting innovative management methodologies. One of the talks will be by Jared Decker from the University of Missouri. The title of his talk is “Matching Cattle Genetics to the Environment Using Genomics.” He will discuss genotype-by-environment genome-wide association analyses his lab has run on cattle in his talk.
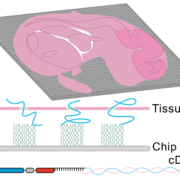
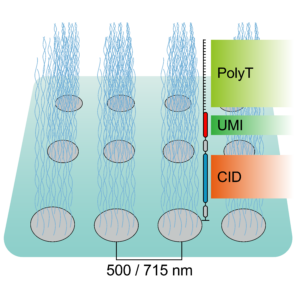
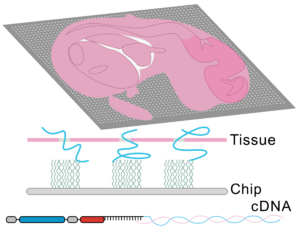
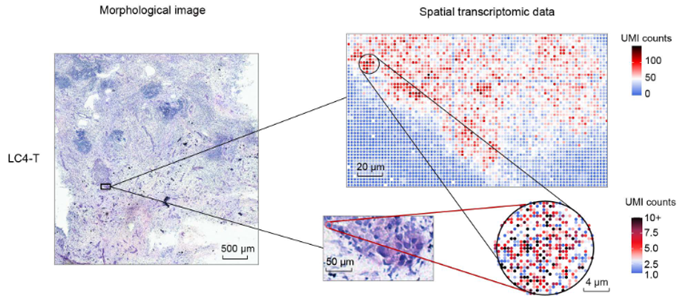
Spatially Resolved Transcriptomics in Plant and Animal Species
New developments in spatial transcriptomics have improved the ability to identify cell types, map cell-fate lineages, and unveil interactions between cells. The ability to characterize gene expression across the entire genome at a subcellular level is crucial for defining single-cell functions and understanding their microenvironments. The strengths and weaknesses of technologies will be discussed. An emphasis on the importance of having complete transcriptome data, as opposed to only having a small panel of genes, will be discussed. The importance of having subcellular resolution will also be addressed. During this session, Mathew Lewsey will give a talk titled “Spatially Resolved Transcriptomic Analysis of the Germinating Barley Grain.” His talk will outline the changes in gene expression during early plant development. This is the phase when the embryo undergoes cellular transitions to become a seedling. Later, during the session, Marc Libault from the University of Missouri will present how his lab uses spatially resolved transcriptomics and metabolomics to understand the cellular complexity in plants.
Day 2, Jan 13, 2024 (Sat)
Single Cell Genomics: This workshop will cover transcriptomics at the cellular level using single-cell analysis techniques. The analysis will be presented using omics approaches and integrating multi-omics data from single cells using novel computational methods. During this session, Gunvant Patil, from Texas Tech University, will present single nucleus RNAseq (snRNAseq) data and gene-editing results to characterize cellular heterogeneity and transcriptional dynamics in soybean leaf. His presentation will focus on the genes and pathways required for the cell-specific accumulation of minerals (ex. Silicon). Also, during this workshop, Maggs X, from Wes Warren’s lab at the University of Missouri, will deliver a talk on a whole brain transcriptome atlas for the blind cavefish (Astyanax mexicanus). Their transcriptome atlas has been supplemented with snRNAseq data from Pachón cave and Rio Choy surface populations. These two populations differ because the Rio Choy population is exposed to sunlight, whereas the Pachón cave population is cave-dwelling and not exposed to sunlight. The single cell data presented will be from over 60,000 cells with 16 broad cell types. Their project aims to shed light on why cave morphs have dysregulated circadian rhythms, reduced aggression, and differences in sensory processing compared to their surface living relatives.
Scott Edwards will give his plenary talk titled “Comparative Population Pangenomes: A New Frontier for the Evolutionary Analysis of Birds.” His lab is investigating how noncoding regions of the genome are driving phenotypic evolution. He will describe how enhancers regulate gene expression to create diverse phenotypes. To accomplish this, his group is developing statistical models that detect evolutionary changes in enhancer sequences that are associated with phenotypic variation within and between species. To validate these statistical models, his lab plans to functionally test how enhancer variations change the development of the forelimb and hindlimbs in birds.
Day 3, Jan 14, 2024 (Sun)
Computational Gene Discovery – This workshop will provide an update on algorithms and software tools (pipelines) for plant and animal genome annotation. Evgenia Kriventseva will discuss the improvements made in ortholog annotation by the new OrthoDB release 11. This latest release provides analysis and annotation for over 100 million genes. Evgenia will also discuss how BUSCO is being used to assess the quality of assembled metagenomes. Also, during this session, Cynthia Webster will introduce EASEL (Efficient, Accurate, Scalable Eukaryotic modeLs). This new genome annotation tool utilizes machine learning, RNA folding, and functional annotations to improve gene prediction accuracy.
Degraded DNA and Paleogenomics – This session will include leading experts in the field of ancient DNA research. In this session, Jonas Oppenheimer will discuss the use of ancient DNA for historical bison populations. Jonas will present evidence from ancient DNA samples to support that bison migrated from Asia across the Bering Land Bridge into the Americas within the past 200,000 years. Once in the Americas, bison separated into two subspecies: wood and plains bison. Later in this session, Beth Shapiro will discuss how genomic data is being used for the hybridization of bison with cattle to create specialized breeds known as cattalo and beefalo. Love Dalen, from Stockholm University, will finish the session with a talk on the biogeography and evolution of North American mammoths. His talk will highlight the gene flow between different mammoth taxa.
Day 4, Jan 15, 2024 (Mon)
Genomics & Biodiversity:
Biodiversity genomics is a rapidly emerging field that captures biological diversity from DNA to ecosystems. Genomics advancements will enhance our understanding of Earth’s biodiversity, promising discoveries vital for present and future societies. This session will discuss how next-generation genomic technologies are being used to describe biological diversity patterns and the mechanisms driving diversification. The topics covered will explore links between genetic variance, biodiversity, and a sustainable world’s health, fostering broader engagement in this exploration.
Day 5, Jan 16, 2024 (Tue)
Functional Annotations of Animal Genomes (FAANG):
The workshop’s primary objectives include:
1) facilitating the sharing of recent FAANG initiatives globally.
2) creating a dynamic platform for the FAANG community to communicate, fostering interactions and collaborations.
During this workshop, Christopher Tuggle, from Iowa State University, will discuss how to assess the success of FAANG and how this can be used for funding the FAANG Farm to Fork agenda. Emily Clark from The Roslin Institute at the University of Edinburgh will provide an update on The H2020 Bovreg Project: An Integrated Functional Annotation of the Bovine Genome. In this talk, she will describe how a diverse dataset, including ChIPseq, ATACseq, RRBS, and WGMS data, was used for the functional annotation of the Bovine Genome. David Hawkins, from the University of Washington, will be giving a similar presentation, but on the functional annotation of the chicken genome.
Bioinformatics: This workshop will focus on the computational tools used for the interpretation of biological data and understanding genomics. This workshop is being led by Aleksey Zimin. During this workshop, Alexandre Lomsadze, from Georgia Tech, will describe the improvements in performance and accuracy of GeneMark-ETP over GeneMark-ET or GeneMark-EP+ for gene predictions. In addition, Haoyu Cheng, from Harvard Medical School, will characterize the HiFiasm assembler. HiFiasm requires PacBio HiFi, ONT ultra-long, Hi-C reads, and trio data. Haoyu will present the results of HiFiasm assemblies from 22 human and two plant genome assemblies.
Galaxy for NGS Data Analysis: This session will be a hands-on workshop that will first introduce the Galaxy platform. The session will also include a hands-on tutorial on the QC, classification, and analysis of metagenomes using short and long-read sequencing data.
Day 6, Jan 17, 2024 (Wed)
Wildlife Genomics: This workshop will discuss the evolutionary and population genomics of wild animals. Aryn Wilder, from the local San Diego Zoo, will present an update on the zoo’s conservation efforts on the Northern White Rhino. Using the southern white rhinos as a benchmark, they are running simulations on the viable fitness of restoring the Northern White Rhino using biobanked cells. Also, during this session, Mikkel-Holger Sinding, from the University of Copenhagen, will be giving a talk on three Asian Bos lineages: gaur (Bos gaurus), banteng (Bos javanicus), and the extinct kouprey (Bos sauveli). Using genomic data from historical samples, his group found that banteng is a paraphyletic group with several Bos species lineages. He suggests that these lineages are a result of significant admixture events.
Keywords: Bioinformatics, Population Genomics, Conservation Genomics, Spatial Biology, Single Cell Genomics, Spatial transcriptomics, Multi-omics, Paleogenomics, Ancient DNA, Agrigenomics, Computational Biology.